
 24 May 2023
24 May 2023

Penetration testing for medical devices: Compliance guide
25 April 2026
 10:55 MIN Read time
10:55 MIN Read time  BY
Amju
BY
Amju
Penetration testing used to be something medical device companies scheduled after launch, if they thought about it at all. That calculus has changed completely. Since October 2023, the FDA has been refusing to accept premarket submissions that lack adequate cybersecurity evidence, including penetration testing results. CDSCO in India and ADHICS in the UAE are tightening their own frameworks in parallel. For CTOs and founders building connected medical devices, this means penetration testing is now a hard regulatory requirement, not an optional security exercise. Get it wrong, and you face submission refusals, delayed market entry, and real patient safety risk.
Table of Contents
- Global regulatory landscape for medical device penetration testing
- What must be penetration tested: Attack surface and methodologies
- Integrating penetration testing into the secure development lifecycle
- Audit, documentation, and handling regulatory edge cases
- A fresh perspective: Why most medical device companies fail on first penetration testing submission
- Next steps: Secure and compliant penetration testing for your medical devices
- Frequently asked questions
Key Takeaways
| Point | Details |
|---|---|
| Regulatory-driven pen testing | FDA, CDSCO, and IEC standards mandate comprehensive penetration testing for medical device compliance. |
| Comprehensive attack surface coverage | Testing must include network, firmware, hardware, wireless, and app components for full vulnerability evaluation. |
| Lifecycle integration essential | Penetration testing should be embedded in the secure development framework and repeated annually or after changes. |
| Audit readiness | Regulatory submissions require detailed documentation, current results, and patient safety risk scoring—not just CVSS data. |
| Avoid common mistakes | Missing documentation, outdated tests, and ignored edge interfaces are leading causes of compliance failures. |
Global regulatory landscape for medical device penetration testing
Regulatory bodies across the US, India, the EU, and the UAE have converged on one clear message: cybersecurity must be built in, tested, and documented before a device reaches patients.
The FDA sets the clearest standard. FDA premarket submissions for cyber devices must include penetration testing evidence as part of the cybersecurity testing package. This applies to any device with software that connects to networks, other devices, or the internet. The FDA’s guidance specifies scope, methodology, independence of testers, and the age of results. Submissions missing this evidence are placed on hold, a process that can delay approval by months.
In India, CDSCO references IEC 81001-5-1 for cybersecurity in medical device software, requiring secure development practices, threat modeling, vulnerability scans, and penetration testing. Auditors verify that these controls exist and that documentation supports them. The EU MDR similarly mandates cybersecurity as part of general safety and performance requirements, and the UAE’s ADHICS framework adds regional specificity for health information systems operating in the Emirates.
Two international standards unify expectations across all these jurisdictions. IEC 81001-5-1 covers health software and health IT systems security, while IEC 62443 addresses industrial automation and control systems, which applies directly to networked medical devices. Together, they define what a medical device security guide should look like in practice.
What do auditors and reviewers actually want to see? The documentation must show:
- Scope: every component, interface, and communication pathway tested
- Methodology: black, grey, or white box approach and tools used
- Independence: who conducted the test and their relationship to the development team
- Results: findings, severity ratings, and remediation status
Healthcare ransomware attacks rose 30% in 2025, and FDA refusals for missing cybersecurity documentation have become routine since October 2023. The cost of skipping penetration testing is no longer theoretical.
The table below summarizes how each jurisdiction treats penetration testing requirements:
| Jurisdiction | Governing body | Standard referenced | Pen test requirement |
|---|---|---|---|
| United States | FDA | FDA Cybersecurity Guidance | Mandatory for premarket submission |
| India | CDSCO | IEC 81001-5-1 | Required via audit verification |
| European Union | EU MDR | IEC 62443, EN standards | Part of safety/performance evidence |
| UAE | ADHICS | ADHICS framework | Required for health IT systems |
Companies in India and the UAE building devices for export to the US or EU face a double compliance burden. The good news is that VAPT for compliance conducted to FDA standards generally satisfies CDSCO and ADHICS requirements as well, since all three reference the same underlying IEC standards.
What must be penetration tested: Attack surface and methodologies
With global requirements mapped, the next step is defining the technical scope for penetration tests.
Medical devices have a far larger attack surface than most software products. A connected infusion pump, for example, might expose a web interface for clinicians, a Bluetooth connection for a mobile app, a USB port for firmware updates, and a hospital network connection for data transmission. Each of these is a potential entry point for an attacker.
The full attack surface for medical device penetration testing includes:
- Network services: open ports, protocols, and communication channels between the device and hospital networks or cloud backends
- Software and firmware: the operating system, application code, and update mechanisms
- Hardware interfaces: USB ports, JTAG debug ports, serial interfaces, and SD card slots
- Wireless protocols: Bluetooth, Bluetooth Low Energy, Zigbee, Wi-Fi, and proprietary medical wireless protocols
- Authentication and access control: login mechanisms, session management, and privilege escalation paths
- Web and mobile applications: companion apps, clinician dashboards, and remote monitoring portals
Hardware debug ports deserve special attention. JTAG and UART interfaces are often left active in production firmware because development teams forget to disable them. An attacker with physical access can use these ports to extract firmware, modify device behavior, or gain root-level access. This is one of the most commonly missed surfaces in first-time submissions.
Wireless protocols used in medical settings also carry unique risks. Bluetooth Low Energy implementations in glucose monitors and cardiac monitors have historically contained pairing vulnerabilities. Zigbee networks used in hospital automation can be intercepted. Testing these requires specialized equipment and expertise beyond standard IT penetration testing.

The choice of testing methodology matters significantly. Here is how the three main approaches compare for medical devices:
| Methodology | Tester knowledge | Best use case | Regulatory preference |
|---|---|---|---|
| Black box | No prior knowledge | Simulates external attacker | Good for network/app layers |
| Grey box | Partial knowledge (architecture docs) | Simulates insider or supply chain threat | Preferred for most device submissions |
| White box | Full source code and design access | Finds deepest logic flaws | Required for firmware and safety-critical code |
Most regulatory submissions benefit from a grey box or white box approach for firmware and a black box approach for network and application layers. This combination gives auditors confidence that both external and internal threat scenarios have been covered.
One critical constraint: all testing must be non-destructive. Unlike IT systems where a crash can be rebooted in minutes, a medical device failure during testing could compromise patient safety data or require hardware replacement. Testers must fix device security gaps without triggering device failures that would invalidate the test environment.
Pro Tip: Always explicitly list every hardware interface in your test scope document, including interfaces you believe are disabled. Auditors will ask, and “we didn’t think it was active” is not an acceptable answer.
The IoT device testing services required for medical devices go well beyond standard web application testing. Make sure your testing partner has specific experience with embedded systems and medical wireless protocols before engaging them.
Integrating penetration testing into the secure development lifecycle
Once you know what to test, it’s vital to understand how to weave penetration testing into your product lifecycle for compliance.
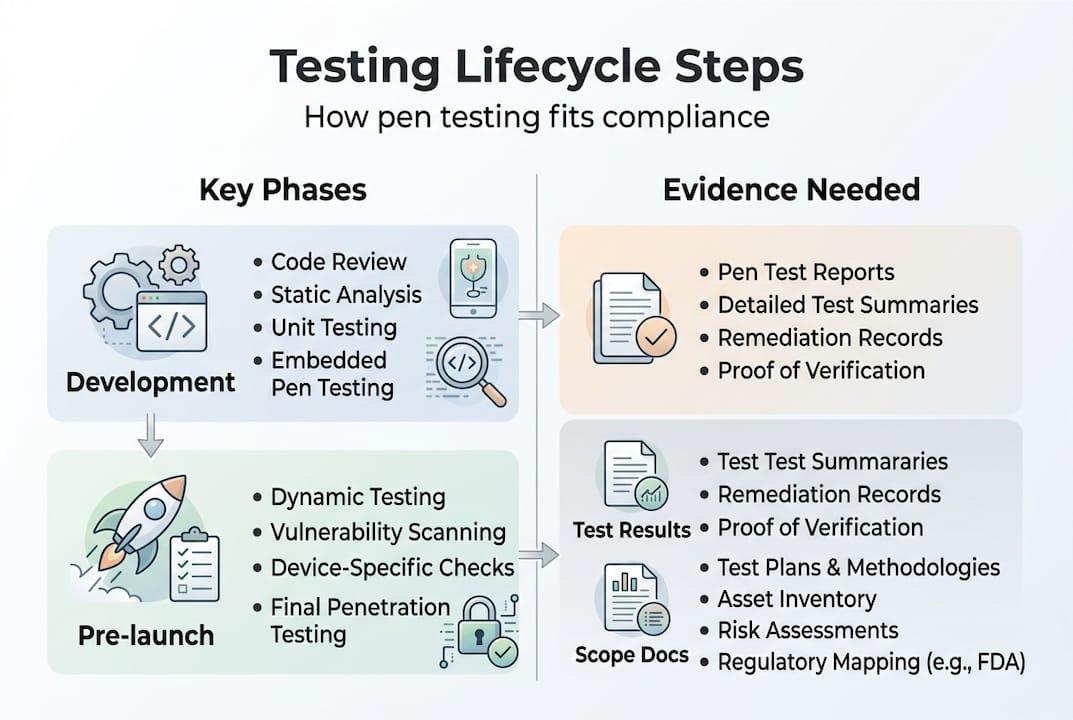
The FDA expects penetration testing to be performed on the final software configuration, meaning the exact firmware and software version you intend to ship. Testing an earlier build and submitting those results is a common mistake that leads to hold letters. If you update firmware after testing, even for a minor bug fix, you need to assess whether a retest is required.
Timing matters. FDA guidance indicates that penetration test results should be less than one year old at the time of submission. Results older than 12 months are considered stale and will not satisfy reviewers. For devices with long development cycles, this means scheduling the penetration test close to the submission date, not at the start of development.
Integrating penetration testing into your Secure Product Development Framework (SPDF) means it is not a one-time event. The SPDF lifecycle connects threat modeling, SBOM (Software Bill of Materials) management, and penetration testing into a continuous loop.
Here is how to structure compliant penetration test submissions:
- Complete threat modeling using STRIDE (Spoofing, Tampering, Repudiation, Information Disclosure, Denial of Service, Elevation of Privilege) before the penetration test begins. This defines what scenarios testers should prioritize.
- Generate and maintain your SBOM so testers know every third-party component in the device. Known vulnerabilities in open-source libraries are a common finding.
- Lock the final software configuration before scheduling the test. No code changes after this point without reassessment.
- Engage an independent or external tester. The FDA strongly prefers testers who have no involvement in device development.
- Document findings with severity ratings using both CVSS scores and patient safety impact scoring.
- Remediate critical and high findings before submission. Document the remediation for each finding, including evidence of retesting.
- Archive all test artifacts: scope documents, test plans, raw findings, remediation evidence, and the final report.
Pro Tip: Build your documentation as testing progresses, not in a rush before submission. Auditors can tell when documentation was assembled after the fact, and it raises questions about the rigor of the process.
One area where many teams stumble is the FDA cybersecurity mistakes that come from treating penetration testing as a checkbox rather than a technical process. Scheduling a test without prior threat modeling, for example, means testers have no prioritized attack scenarios to work from. The result is a report that looks thorough but misses the vulnerabilities that matter most for patient safety.
Audit, documentation, and handling regulatory edge cases
Finally, you need to prepare for rigorous audits, navigate nuances, and avoid the mistakes that trip up most applicants.
Auditors reviewing medical device cybersecurity submissions look for four qualities: completeness, currency, scope, and evidence of methodology. A report that covers only the web interface while ignoring firmware and hardware ports will fail on completeness. report from 18 months ago will fail on currency. This report that lists findings without explaining how they were discovered will fail on methodology evidence.
Patient safety risk scoring is a critical differentiator from standard IT security. CVSS scores alone are insufficient for medical device submissions. A vulnerability rated CVSS 5.0 might be low priority in an enterprise IT context but critical for a device that delivers medication doses. Auditors expect to see patient safety impact scoring layered on top of CVSS, explaining how each vulnerability could affect device function and patient outcomes.
FDA refusals since October 2023 have consistently cited missing or incomplete cybersecurity documentation. Healthcare ransomware is up 30% in 2025. These are not abstract risks. They represent real submission delays and real patient harm.
CDSCO audits in India verify that IEC 81001-5-1 controls are implemented across the full development lifecycle. This includes secure updates, vulnerability management, and penetration testing. Auditors will ask for evidence of each control, not just a declaration that it exists.
Here is a practical audit preparation checklist:
- Confirm the penetration test was performed on the final software configuration
- Verify the test report is less than 12 months old at submission date
- Check that all attack surfaces are explicitly listed in the scope section
- Confirm tester independence is documented (no affiliation with the development team)
- Ensure all critical and high findings have documented remediation and retest evidence
- Include patient safety impact ratings for every finding
- Archive the SBOM version used during testing
- Document any post-test changes and their impact assessment
When devices undergo significant updates, the compliance audit security process must restart for affected components. A firmware update that changes network communication protocols, for example, requires retesting of all network-layer findings. Many companies miss this and submit outdated results for updated devices, which triggers hold letters.
The compliance management case for building a living documentation system, rather than a static report, is strong. When auditors ask follow-up questions months after submission, you need to be able to answer them with current evidence, not scramble to reconstruct what happened during testing.
A fresh perspective: Why most medical device companies fail on first penetration testing submission
Most teams that fail their first penetration testing submission are not careless. They are experienced engineers who treated penetration testing like software QA: schedule it near the end, find the bugs, fix them, done. That mental model does not work for regulatory submissions.
The real problem is timing and documentation culture. Companies wait until they receive an FDA hold letter before taking penetration testing seriously. By then, they have lost months of market entry time and often need to retest a device that has already gone through multiple firmware iterations.
From working with medical device teams, the most common blind spots are hardware debug ports left active in production builds, medical wireless protocol testing skipped because “we use a certified radio module,” and threat modeling done after penetration testing instead of before. Each of these produces a submission that looks complete but fails under scrutiny.
The lessons from FDA-ready device testing point to one consistent improvement: treat penetration testing as a continuous process with documentation built in real time. Not a one-off event before submission.
Practical improvements that make a real difference:
- Start threat modeling at the architecture phase, not after development
- Assign a documentation owner who captures test artifacts as they are created
- Schedule penetration testing 60 to 90 days before your target submission date
- Retest after every significant firmware change, even minor ones
Next steps: Secure and compliant penetration testing for your medical devices
Regulatory requirements for medical device penetration testing are specific, time-bound, and unforgiving of gaps. Independent VAPT conducted to FDA, CDSCO, and IEC standards is not something to improvise close to a submission deadline.

Testvox specializes in medical device security testing that is built for regulatory submissions, not just internal audits. Our security testing services cover the full attack surface, from firmware and hardware interfaces to wireless protocols and companion apps, with documentation structured for FDA, CDSCO, and ADHICS review. Whether you need a first-time submission package or a retest after a firmware update, our VAPT testing solutions deliver audit-ready results. Reach out to Testvox to start building a penetration testing program that keeps your device on track for regulatory approval.
Frequently asked questions
How often should penetration testing be performed on medical devices?
Penetration testing should be repeated annually or whenever major software changes occur. FDA accepts test results less than one year old at the time of premarket submission.
What evidence is required in compliance submissions for penetration testing?
Compliance submissions must include the test scope, methodology used, tester independence documentation, detailed findings, and remediation evidence. FDA premarket submissions for cyber devices require all of these elements as part of the cybersecurity testing package.
What attack surfaces should be covered during pen testing for medical devices?
The full attack surface includes network services, software and firmware, hardware interfaces such as USB and JTAG, wireless connections, authentication mechanisms, and web or mobile application layers.
What standards should medical device pen testing comply with in India and UAE?
Medical device penetration testing should follow IEC 81001-5-1 and IEC 62443, which support CDSCO audit requirements in India and align with the ADHICS framework in the UAE, as well as FDA and EU MDR expectations.
Recommended
- VAPT Archives – Testvox
- Medical Device Security Testing: A Complete Guide
- Security Testing Case Study: Compliance management system – Testvox
- Securing a Ticket Booking Application with VAPT – Testvox
24 May 2023
 27 November 2025
27 November 2025


ABOUT TESTVOX

Testvox is a software testing company help your product reach its full potential. Get full cycle testing for your mobile and web applications while ensuring all quality assurance standards are met... Read More
100+
Applications
Assured
Assured
5 Star Rating
5 Star Rating
“By participating in our release lifecycle, Testvox's standard QA process has helped us to launch quality products faster.”

Rima Hudaifa
Manager | Cadvil
Manager | Cadvil
