





UAE
Testvox FZCO
Fifth Floor 9WC Dubai Airport Freezone

 29 April 2026
29 April 2026
 9:33 MIN Read time
9:33 MIN Read time  BY
Amju
BY
Amju
Most medical device founders assume that if their device works reliably and safely, regulators will wave it through. That assumption is costing startups months of delays and, in some cases, complete market rejections. Cybersecurity documentation is no longer a footnote in your regulatory submission. For India’s CDSCO, the UAE’s MOHAP, and the FDA, it is a core gatekeeper requirement sitting alongside clinical evidence and device safety data. This guide gives you a clear, actionable roadmap to align your product’s cybersecurity posture with what regulators actually expect, so your launch stays on schedule.
| Point | Details |
|---|---|
| Cybersecurity as a gatekeeper | Market access for medical devices increasingly depends on meeting cybersecurity requirements, not just core functionality. |
| Global regulatory alignment | Adhering to IEC 81001-5-1 and preparing required evidence streamlines approval in India, UAE, and beyond. |
| Integrate early, not late | Embedding security into your QMS and SDLC from day one avoids launch delays and compliance gaps. |
| Local nuances matter | India and UAE have unique twists—such as VAPT and ADHICS—that require country-specific attention along with global best practices. |
| Continuous vigilance | Launch is just the start—ongoing patching, disclosure, and security assessments protect both market access and patient safety. |
The regulatory landscape shifted decisively over the past three years. Cybersecurity incidents involving connected medical devices triggered real-world patient harm events, and regulators responded by making cybersecurity evidence a mandatory part of premarket review, not an optional supplement. Today, FDA requires cybersecurity documentation in premarket submissions for any device carrying cybersecurity risks, which effectively means any device with software, wireless connectivity, or network access.
“A cybersecurity gap in your submission is no longer a minor deficiency. It can trigger a complete regulatory hold, forcing a full re-submission cycle.”
The consequences of non-compliance are severe. Regulatory holds delay your time-to-market by six to eighteen months on average. Market withdrawals damage your brand with distributors and hospital procurement teams. In some cases, devices are banned outright from import. These are not theoretical risks. They are documented outcomes that Indian and UAE startups have already experienced when entering FDA or EU markets without adequate cybersecurity evidence.
In the UAE, MOHAP regulates medical devices and increasingly aligns its expectations with EU MDR and IEC 81001-5-1 (the global standard for health software cybersecurity). India’s CDSCO has similarly tightened its documentation requirements, particularly around software-based devices. The practical takeaway: if you are building toward any of these markets, you need a cybersecurity compliance strategy from day one. Reviewing FDA cybersecurity mistakes that other companies have made is a useful starting point for understanding where submissions typically break down.
Key risks to keep front of mind:
Understanding what regulators actually want is where most startups get lost. The requirements are not vague. They are specific, document-heavy, and interconnected. Here is what you need to assemble.
| Market | Key framework | Core documents required |
|---|---|---|
| USA (FDA) | SPDF, IEC 81001-5-1 | SBOM, cybersecurity management plan, threat model, VAPT report, CVD policy |
| India (CDSCO) | National guidelines, VAPT | Risk file, VAPT evidence, software documentation |
| UAE (MOHAP) | EU MDR, IEC 81001-5-1, ADHICS | Conformity evidence, cybersecurity risk assessment, facility compliance |
| EU (MDR) | IEC 81001-5-1, GDPR | Technical file with cybersecurity annex, postmarket surveillance plan |
FDA requires an SBOM, cybersecurity management plans, and Secure Product Development Framework (SPDF) evidence with risk-based justification. An SBOM (Software Bill of Materials) is essentially an ingredient list for every software component in your device, including third-party libraries and open-source packages. Regulators use it to assess whether known vulnerabilities in those components have been addressed.
For India and the UAE specifically, VAPT plus a risk file is the baseline expectation for CDSCO, while UAE regulators expect global alignment with IEC 81001-5-1, EU MDR, and ADHICS for device-handling facilities. ADHICS (Abu Dhabi Healthcare Information and Cyber Security Standard) adds a facility-level layer that many startups miss entirely when planning UAE market entry.
There is also a critical risk of over-regulation consequences when documentation is ignored. Aggressive enforcement is not a future possibility. It is current practice.
Postmarket requirements deserve special attention. Even if you are launching with limited distribution, you need a coordinated vulnerability disclosure (CVD) policy and a documented patching plan. Regulators want to see that you have a process for responding to vulnerabilities discovered after launch, not just before it.
Pro Tip: Adopt IEC 81001-5-1 as your baseline framework even if your primary market is India or the UAE. It is recognized across all major regulatory jurisdictions, which means one harmonized framework reduces the documentation burden when you expand internationally.
For a deeper breakdown of what penetration testing compliance looks like in practice, and to see a real-world example through a security testing for FDA submission, these resources give you concrete context.
Most startups treat cybersecurity compliance as a pre-submission task. That is the single most expensive mistake you can make. By the time you are assembling your regulatory package, it is too late to retrofit security into your architecture without significant rework.
IEC 81001-5-1 integrates with existing SDLC without requiring a complete overhaul. Embedding it into your Quality Management System (QMS) early prevents the delays that come from discovering gaps at submission time. Here is how to do it in practice:
Focusing on authentication and code security prevents up to 60% of vulnerabilities, and maturity frameworks consistently raise compliance scores across organizations that adopt them systematically.
“Security is not a feature you add at the end. It is an architecture decision you make at the beginning.”
Secure-by-design principles, including defense-in-depth strategies covering authentication, encryption, and active monitoring, are explicitly called out in FDA’s QMS guidance. Cross-team collaboration is essential here. Cybersecurity cannot be one engineer’s responsibility. It needs to be embedded in your product, clinical, regulatory, and software teams simultaneously.

Pro Tip: Assign a cybersecurity owner to your QMS who participates in design reviews, not just security reviews. This person bridges the gap between regulatory documentation and engineering reality.
Keep an eye on security testing trends that are reshaping how regulators evaluate submissions, and review VAPT case studies to understand what a compliant testing engagement actually produces.
Both markets have specific nuances that generic compliance guides miss entirely.
UAE-specific considerations:
India-specific considerations:
Connectivity risks that trip up both markets:
Threat modeling for legacy devices and Bluetooth connectivity is an area of increased regulatory scrutiny. Home-use products face even higher examination because the network environment is uncontrolled. If your device connects to a cloud backend, uses Bluetooth for patient data transfer, or integrates with legacy hospital systems, each of those connections is a separate attack surface requiring documented risk analysis.
| Challenge | India (CDSCO) | UAE (MOHAP/ADHICS) |
|---|---|---|
| VAPT requirement | Mandatory for software devices | Expected, aligned with EU MDR |
| Connectivity risk documentation | Risk file required | IEC 81001-5-1 threat model |
| Postmarket surveillance | Required | Required, facility-level ADHICS |
| Legacy device integration | Case-by-case review | Higher scrutiny |
Strategies for fixing device security gaps before submission and a practical security compliance case study illustrate how these regional challenges translate into real testing and documentation work.
Here is the step-by-step sequence that takes you from early development to regulatory submission without last-minute scrambles.
Premarket documentation including SBOM, CVD, and testing evidence is required for compliance across all major markets. Ongoing patch management is equally mandatory, not optional.
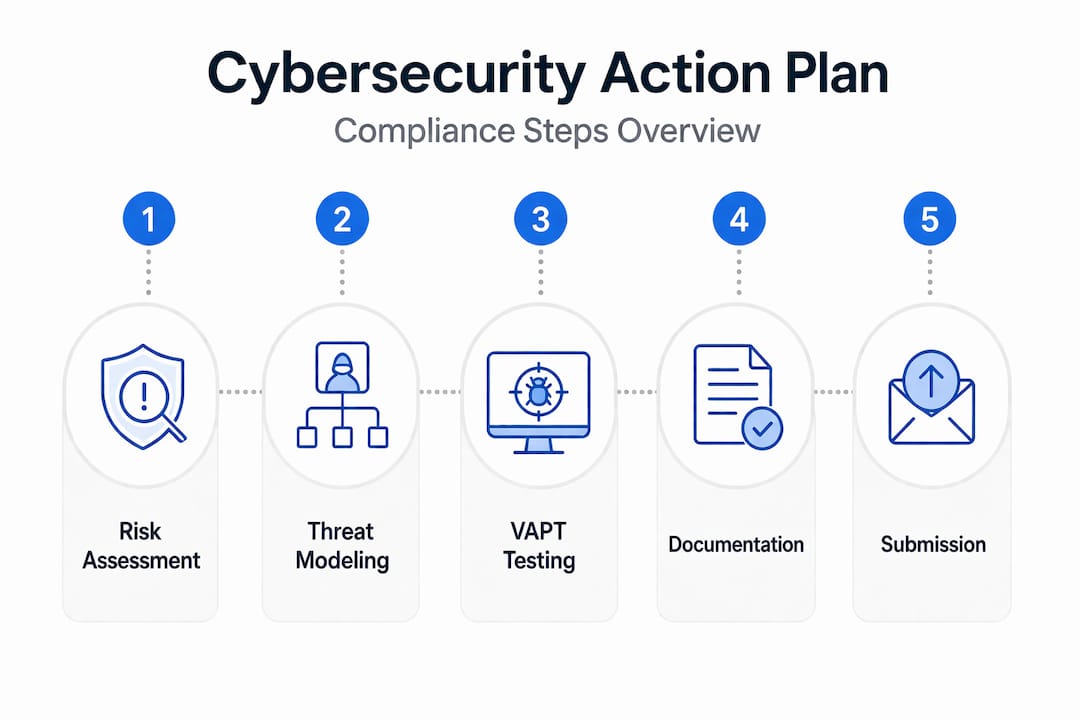
Pro Tip: Start compliance tasks in parallel with design work. Startups that treat compliance as a sequential step after engineering spend two to three times more on rework than those who run both tracks simultaneously.
For a detailed breakdown of the penetration testing steps that belong in your submission package, the process is more structured than most founders expect.
We see the same pattern repeatedly. A startup builds a genuinely innovative device, rushes through development, then spends three months trying to reverse-engineer compliance documentation before a regulatory deadline. The documentation looks like compliance. It is not. Regulators can tell the difference between security evidence that emerged from a real secure development process and a document that was written to fill a gap.
The deeper problem is treating cybersecurity as a regulatory box to check rather than a design imperative. When security is bolted on at the end, the patches are fragile, the threat models are incomplete, and the disclosure processes are theoretical. Regulators are getting better at identifying this pattern, and enforcement is tightening.
Regulatory requirements will keep rising. Compliance alone is no longer a competitive differentiator because your competitors will eventually meet the same bar. What differentiates successful launches is how early and how deeply security is embedded into the product culture. Startups that automate security checks, run continuous vulnerability reviews, and maintain living threat models sidestep the last-minute rush entirely.
The mindset shift is this: stop asking “what do we need to document?” and start asking “what risks does our device actually carry, and how do we manage them transparently?” That question leads to better products, faster submissions, and fewer postmarket surprises. Addressing device cybersecurity gaps proactively is what separates teams that launch on schedule from those that spend a year in regulatory limbo.
Getting your medical device past cybersecurity compliance gates is faster when you work with a team that has done it before across India, UAE, and FDA submissions.

Testvox specializes in comprehensive security testing for medical devices, delivering VAPT reports, SBOM reviews, threat model validation, and full documentation packages aligned with CDSCO, MOHAP, and FDA requirements. Our team works alongside your engineering and regulatory teams, not around them, so compliance evidence is traceable, defensible, and submission-ready. Explore our security testing service to see how we structure engagements for startups at every stage of the product lifecycle.
Key documents include a Software Bill of Materials (SBOM), a cybersecurity management plan, Secure Product Development Framework evidence, and coordinated vulnerability disclosure procedures, all of which FDA requires in premarket submissions.
India requires VAPT and a risk file for CDSCO submission; the UAE aligns with EU MDR/IEC 81001-5-1 and applies the ADHICS standard at the facility level.
IEC 81001-5-1 is a harmonized global standard for medical device cybersecurity that integrates with your QMS; prioritizing IEC 81001-5-1 compliance accelerates multi-market access by satisfying multiple regulatory frameworks simultaneously.
Start cybersecurity integration at the design phase, document threat models and mitigation plans, and prepare coordinated vulnerability disclosures before submitting, because aggressive enforcement leads to project holds when documentation is incomplete.
Maintain a disclosure and patching process tied to your QMS, and regularly review your SBOM and threat models as new vulnerabilities emerge, since postmarket surveillance with coordinated vulnerability disclosure is a standing regulatory requirement.



Let us know what you’re looking for, and we’ll connect you with a Testvox expert who can offer more information about our solutions and answer any questions you might have?